
by Richard Peachey
What is Sickle-Cell Anemia?
Sickle-cell anemia is a genetic disease common to persons of West and Central African ancestry. It is characterized by severe anemia with symptoms of pallor, muscle cramps, weakness, and susceptibility to fatigue. Additional symptoms include heart enlargement, brain cell atrophy, and severe pain in the abdomen, back, head, and extremities (see diagram below). Many victims of sickle-cell anemia die before the age of twenty, though some survive past fifty.
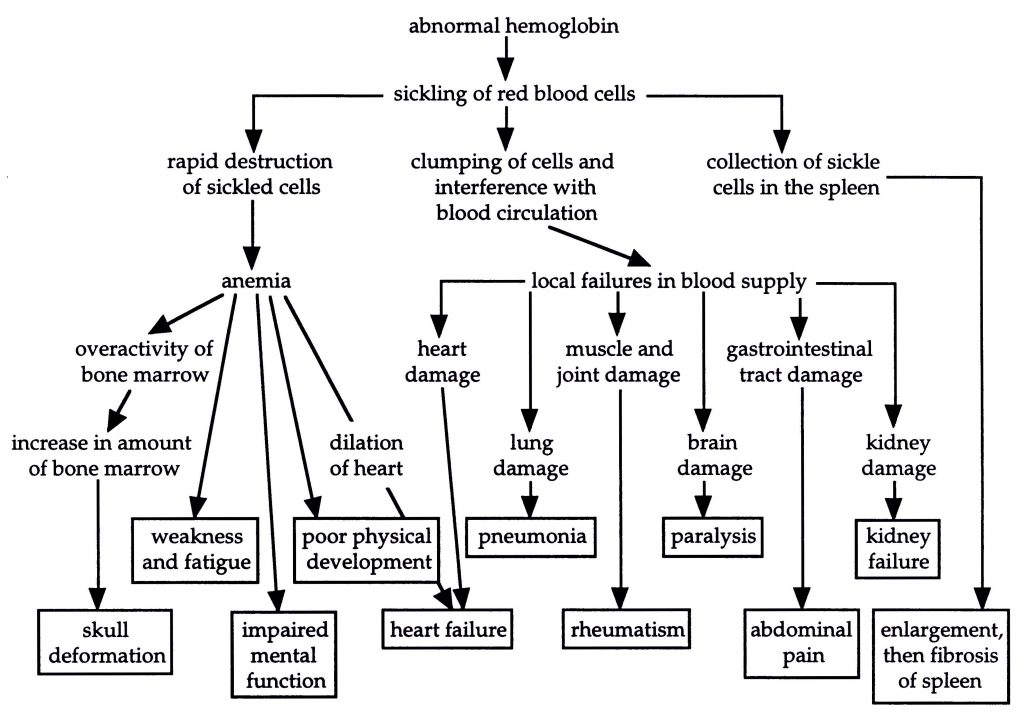
Causes of symptoms seen in sickle-cell anemia victims (based on Starr and Taggart, p. 173)
The symptoms of this disease arise from the tendency of the victim’s red blood cells to “sickle,” that is, to take on irregular shapes. These cells last for only half as long as normal red blood cells; they tend to rupture and are destroyed by the body. In addition, sickled cells often get caught in the capillaries, the narrowest blood vessels, where they clump and produce a blockage. The resulting impaired circulation causes damage to various tissues, in many cases leading to premature death.
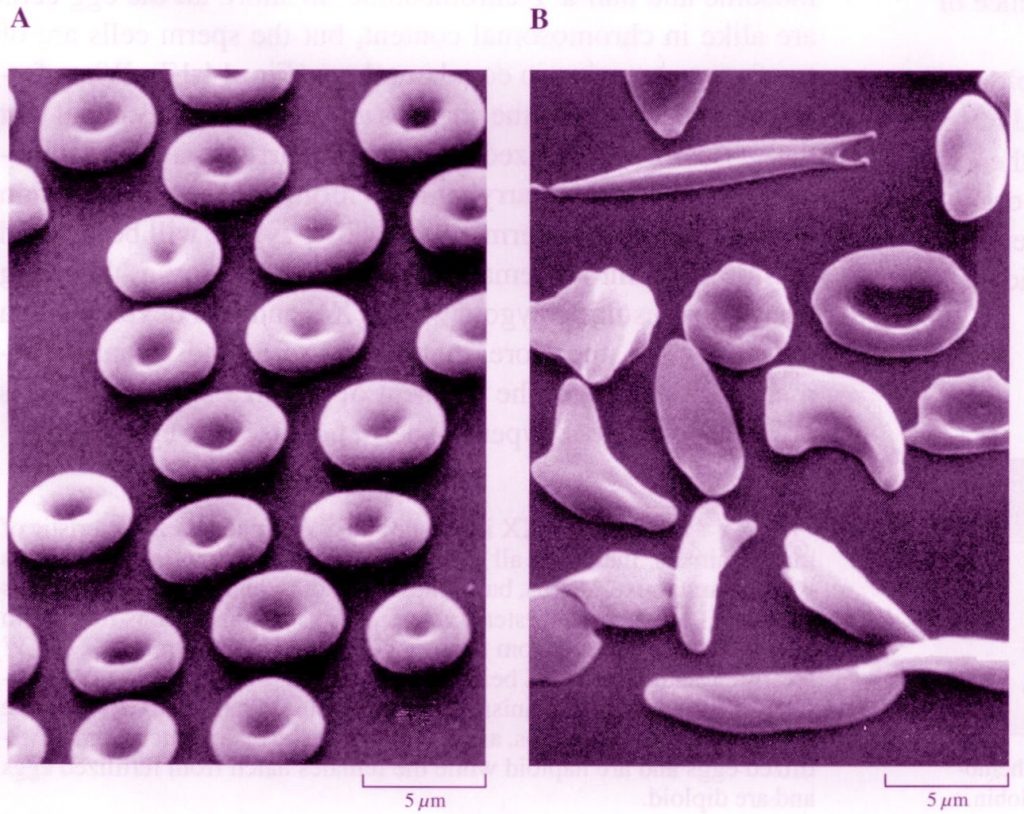
Normal (A) versus Sickled (B) red blood cells (from McFadden and Keeton, p. 293)
What Causes Sickle-Cell Anemia?
The primary function of red blood cells is to carry oxygen from the lungs to all tissues of the body. Each red blood cell contains millions of molecules of hemoglobin, the protein that does the work of oxygen transport. The hemoglobin molecule is composed of four polypeptides (chains of amino acids): two alpha (α) chains, each with 141 amino acids, and two beta (β) chains, each with 146 amino acids. Each of these four polypeptide chains associates with one iron-containing heme group, which has the ability to reversibly bind a single oxygen molecule. Thus the hemoglobin molecule as a whole contains four heme groups and is able to transport four oxygen molecules.
![]()
Hemoglobin molecule (from Price, p. 333, based on Cerami and Peterson)
The cause of sickle-cell anemia is a “point mutation,” that is, the alteration of a single nucleotide base within the DNA of the gene coding for the beta-hemoglobin polypeptide. The sixth DNA triplet, CTC, has been changed to CAC (the nitrogenous base thymine is replaced by adenine in the mutant gene).
This mutation in the beta-hemoglobin gene results in the change of just a single amino acid within the polypeptide encoded by that gene. The sixth amino acid in beta-hemoglobin is normally glutamate, which has an acidic, very polar, “hydrophilic” (water-attracting) side chain. But in the mutant beta-hemoglobin this glutamate is replaced by valine, which has a nonpolar, “hydrophobic” (water-repelling) side chain. This valine at position six in the mutant polypeptide forms a “hydrophobic association” with the nearby valine at position one, causing the abnormal hemoglobin molecule (called “hemoglobin S”) to be less soluble in water than the normal version (called “hemoglobin A”).
As a result of this single altered amino acid in each of the two beta chains, the hemoglobin molecule, when deoxygenated, tends to polymerize and “crystallize” (sometimes irreversibly) into long fibrils, stiff rod-like structures that force the red blood cells containing them to twist into bizarre, jagged shapes. When this occurs, the hemoglobin can no longer load and unload oxygen efficiently; even worse, the red blood cells become distorted and obstruct the capillaries. (Normal hemoglobin has a jelly-like consistency, allowing red blood cells to squeeze easily through the narrowest blood vessels.)
(For more recent information on the biochemical link between malaria and sickle-cell disease, see the Cell articles noted under “further reading,” below.)

Sickle cell anemia — long crystallized fibrils (from Cerami and Peterson, p. 45)
How Can This Be An Example Of A Beneficial Mutation?
Normal individuals have two “good” genes for the beta chain of hemoglobin, one from each parent, in addition to two normal genes for the alpha chain. The genetic trait of normal hemoglobin is designated “HbA,” and the genotype of the individual is written “HbA HbA.” Such people are said to be homozygous for normal hemoglobin; 100% of their hemoglobin will be normal.
Individuals with sickle-cell anemia have two mutant beta-hemoglobin genes; their genotype for hemoglobin is written “HbS HbS.” These people are said to be homozygous for the mutated hemoglobin; all of their hemoglobin will contain valine instead glutamate at the sixth position in each beta chain.
It is possible, however, for a child to inherit one of each version of the beta-hemoglobin gene. This heterozygous genotype is written “HbA HbS.” Such individuals are said to be “carriers” or to have “sickle-cell trait;” their red blood cells produce about 60% normal hemoglobin and 40% mutant hemoglobin. Some of the cells will be sickled, but only 2 to 4% of the level seen in HbS homozygotes. Generally these people lead normal lives, encountering difficulty only in situations of very low oxygen tension, such as at very high altitudes or in situations of extremely strenuous exercise.
The mutation is not beneficial to those who have two copies of the mutated gene in their cells (the HbS homozygotes); they suffer greatly and often die before reaching reproductive age. But heterozygotes (HbA HbS) do receive a benefit: they are less likely to succumb to malaria. Many African infants with normal hemoglobin die of cerebral malaria, but those with sickle-cell trait have greater resistance.
The deadliest form of malaria is caused by the protist Plasmodium falciparum, which enters human blood when the person is bitten by a mosquito (genus Anopheles). The protist pathogen lodges in human red blood cells, but it decreases the pH of the cells by about 0.4 pH units, causing about 40% of infected cells to sickle. These deformed red blood cells are sequestered and phagocytized by immune system cells; thus the protist is destroyed along with the sickled cells. Although this does not provide complete protection from malaria, it does lessen the severity of the disease.
Interestingly, women with sickle-cell trait appear to be more fertile than normal women. The reason for this is not known.
Does This Example Support Macroevolution?
In a limited sense, this mutation can indeed be said to be ”beneficial.” Heterozygotes (HbA HbS) are more likely to live to reproductive age and pass on their genes than are the HbAhomozygotes. They receive significant protection from malaria, and in addition, heterozygote women seem to have greater fertility.
But the mutation is nonetheless a loss of information. The hemoglobin’s normal function is impaired, not improved, and the protection from malaria is simply an incidental side benefit — the pathogen happens to be destroyed along with the person’s own defective cells. This mutation does not introduce a new level of complexity; there is no new functional information or novel structural feature for evolution to build on. Considered in itself, this mutation is destructive and harmful, as are so many others. It is difficult to see how any genetic change of this sort could lead to a true evolutionary advance.
References
Cerami, Anthony, and Charles M. Peterson. 1975. “Cyanate and Sickle-Cell Disease.” Scientific American 232(4):44-50.
Davis, Percival, and Dean H. Kenyon. 1993. Of Pandas and People: The Central Question of Biological Origins. (2nd edition). Dallas: Haughton Publishing Co.
Keeton, William T., and James L. Gould. 1986. Biological Science. (4th edition). New York: W. W. Norton & Co.
Lehninger, Albert L. 1975. Biochemistry: The Molecular Basis of Cell Structure and Function. (2nd edition). New York: Worth Publishers.
McFadden, Carol H., and William T. Keeton. 1995. Biology: An Exploration of Life. New York: W. W. Norton & Co.
McLeod, Kevin Curtis. 1982 (Jun). “The Sickle Cell Trait.” Creation Research Society Quarterly 19:19-26.
Price, Peter W. 1996. Biological Evolution. Orlando, FL: Saunders College Publishing (Harcourt Brace).
Raven, Peter H., and George B. Johnson. 1988. Understanding Biology. St. Louis: Times Mirror/Mosby College Publishing.
Starr, Cecie, and Ralph Taggart. 1989. Biology: The Unity and Diversity of Life. Belmont, CA: Wadsworth Publishing Co.
For further reading:
“Sickle cell anemia does not prove evolution!” by Felix Konotey-Ahulu, a world authority on sickle-cell disease <http://creation.com/sickle-cell-anemia-does-not-prove-evolution>
“An Antioxidant Link between Sickle Cell Disease and Severe Malaria” by Ashraful Haque and Christian R. Engwerda, Cell, Vol. 145 (Apr 29, 2011), pp. 335f. This brief article previews a longer technical report in the same issue, “Sickle Hemoglobin Confers Tolerance to Plasmodium Infection” by Ana Ferreira et al. (pp. 398-409).